Translate this page into:
Clinical Trial - An Introduction to Essential Prerequisites
Address for correspondence: The Editor/ Managing Editor, Journal of Comprehensive Health Dept of Community medicine NRS Medical College, 138, AJC Bose Road, Kolkata-700014
Corresponding Author: Dr. Bobby Paul Postal address: P-19 Jadavpur University Employees’ Housing Co-operative Society Ltd. P.O.- Panchasayar. Kolkata 700 094. West Bengal. India. E-mail : drbobbypaul@gmail.com , dr_bobbypaul@rediffmail.com, Phone no.: +91 98300 46821
INTRODUCTION
In today’s global scientific era, clinical trials are the mainstay for bringing newer and better drugs to market and it has become an indispensable part of the drug discovery process to ensure safety and efficacy of any new drug. According to the Associated Chambers of Commerce and Industry, India is set to grab clinical trials business valued at approximately US$ 1 billion by 2010, up from US$ 200 million last year, making the subcontinent one of the world’s preferred destinations for clinical trials. Drug companies are drawn to India for several reasons, including a techni-cally competent workforce, patient availability, low costs and a friendly drug-control system which is good news for Indian economy. On the contrary, the booming clinical trial industry is raising concerns because of lack of regulations regarding trials by private drug companies, uneven application of protocol for informed consent and proper ethical review 1.This article tries to give a general overview regarding different steps and criteria of conduction of clinical trials.
Clinical trial:
Clinical trial is an organized research, conducted on human beings to investigate the safety and efficacy of a drug. Clinical trial must conform to the moral and scientific principles that justify medical research and should be based on laboratory and animal experiments or other scientifically established facts, and it should be conducted only by scientifically qualified persons and under the supervision of a qualified medical man.
New drug development process:
Drug discovery requires two basic steps i.e. research and development. The research phase consists of three sequential activities comprising of target selection, drug selection and product development. The first phase, target selection, involves choosing a disease to treat and then developing a model for that disease. The second phase, drug selection, is a process that involves finding a drug or group of drugs that work within that model system. Typically the screening process involves hundreds of compounds that are tested against the target. When the compounds with the desired activity are discovered, the most promising among them are optimized to produce one or two final compounds that may eventually become drugs. The entire process from target selection to product development usually takes at least 3 years, and can involve hundreds of researchers and millions of dollars. Then comes the drug development process, which consists of preclinical and clinical development (clinical trial).
Preclinical Development:
Preclinical tests are performed in the laboratory, using a wide array of chemical and biochemical assays, cell-culture models and animal models. This preclinical testing develops pharmacological profile of the drug, determines acute toxicity of the drug in at least two species of animals and conducts short term toxicity studies. Animal pharmacology testing is performed in animal models of human disease. If the result of the toxicity studies indicates that the range between biological activity and toxicity is acceptable, the company will apply to the regulatory body for permission to continue development. The ultimate aim of preclinical testing is to assess safety & biological activity in animals. This phase takes about approximately 4 years. After completing preclinical testing, the company files an Investigational New Drug application to begin to test the drug in human. If regulatory body does not disapprove it within 30 days, then this application becomes effective, and at this stage clinical trial can be initiated. Clinical trials on patients in different countries are approved and monitored by different regulatory agencies, which, in India, is monitored by Drug Controller of India (DCGI) under Central Drug Standard Control Organization (CDSCO). The total duration of these trials may stretch to approximately 8 - 9 years. Clinical research is done in four phases (I, II, III and IV), each designed to address different issues. The knowledge gained from one phase is utilized in the subsequent phases.
Phase I trial (Human Pharmacology):
The main objective of this phase is to establish initial safety, maximum tolerance and pharmacokinetics of the drug in humans. This phase is usually carried on 20-80 healthy human volunteers or certain types of patients. The time period being approximately 3 - 6 months. This phase is also known as ‘First in Man’. In case the drug to be investigated has a known potential adverse effect, the study is carried on upon subjects only for whom the drug is targeted e.g. anticancer drugs are never tested in healthy volunteers, rather it is investigated on cancer patients. Only about 70% of experimental drugs pass phase I trial.
Phase II trial (Therapeutic Exploratory Trials):
On completion of the Phase I trial, the Phase II trial is initiated to evaluate the safety and efficacy of the drug. This phase is conducted on patients either in an open, non b5t6lind or as placebo control, blinded trials. Here 100 - 300 patients are enrolled to determine the dose and adverse reactions of the drugs. This phase may last from 6 months to two years. This phase is further divided into two subtypes - Phase Ila and Ilb.
Phase IIa: Pilot clinical trials to evaluate safety in selected patient population (dose response, type of patient etc.)
Phase IIb: Controlled clinical trials to evaluate safety as well as efficacy for determining a dose range to be studied in phase-III. Only about 35% of experimental drugs pass phase II trial.
Phase III trial (Therapeutic Confirmatory Trials):
This phase involve several hundred to several thousand patients and may last for 1 - 5 years conducted in multicentric manner. Drugs safety and effectiveness are being studied in different patient subgroups like children, elderly and patients having hepato-renal impairment. Once this phase III trial has been completed successfully, the drug company is in a position to apply to the regulatory authorities for marketing approval. This phase again is of two subtypes - IIIa and IIIb.
Phase IIIa: Conducted after the drug’s efficacy is demonstrated but before the regulatory submission of New Drug Application (e.g. studies in children, patient with renal dysfunction etc.) Phase IIIb: Conducted after regulatory submission but prior to the drug’s approval or launch (e.g. to supplement or complement earlier trials). Only about 25% of experimental drugs pass through phase III trial
Phase IV trial (Post Marketing Studies/Trials):
Post marketing trials are studies (other than routine surveillance) performed after drug approval and related to the approved indications. After the prior demonstration of the drug’s safety, efficacy and dose definition in the previous trials, this phase is concerned with the application of the drug in general population. Drug may be withdrawn from the market if some notorious side effects are noticed in phase IV trial, just what happened in Thalidomide tragedy in 1962 2 New Drug Development is a very long journey, as it takes 12 years and ~ 800 million US $ to bring one new drug to market. To start with 5,000-10,000 compounds are initially screened in pre-clinical development, subsequently 250 enter in pre-clinical testing and 5 enter in clinical testing, ultimately 1 compound is approved by the Food and Drug Administration of USA (FDA).
Schedule Y:
Requirements and guidelines for permission to import and or manufacture of new drugs for sale or to undertake clinical trials are mentioned here. Application for permission to import or manufacture new drugs for sale or to undertake clinical trials are made in Form 44 with data of chemical, pharmaceutical information, animal pharmacology data (that includes specific pharmacological actions, general pharmacological actions and pharmacokinetic data), animal toxicology data, human clinical pharmacology data, regulatory status of the drugs in other countries, complete testing protocols and indication of drugs, purpose of examination and application for import of small quantities of drugs.
Regulatory Framework:
Before a clinical trial is initiated it is imperative to follow international ethical and scientific quality standard for designing, conducting, recording and reporting of clinical trials that involve participation of human subjects. Compliance with this standard provides assurance that the rights, safety and well being of trial subjects are protected, consistent with the principles laid down by Declaration of Helsinki and the data are credible 3,4. In India, the regulatory framework is governed by these following guidelines i.e.
i. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human use - Good Clinical Practice: consolidated guideline (ICH-GCP, 1997) 5,
ii. Local Regulatory Requirements (DCGI),
iii. Indian Council of Medical Research (ICMR code, 2000)6
iv. Local Ethics Review Boards (ERBs).
Good Clinical Practice:
Good Clinical Practice (GCP) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety and well-being of trial subjects are protected, consistent with the principles that have their origin in the declaration of Helsinki and the clinical trial data are credible. The guideline was developed with consideration of the current good clinical practices of the European Union, Japan and the United States, as well as those of Australia, Canada, the Nordic countries and the World Health Organization (WHO). This guideline should be followed while generating clinical trial data that are intended to be submitted to regulatory authorities. The principles established in this guideline may also be applied to other clinical investigations that may have an impact on the safety and well-being of human subjects. Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks.
Rationale of Good Clinical Practice:
i. Legal Requirement for conduction of the trial
ii. Protects the rights, integrity & confidentiality of research subjects
iii. Provides assurance that the data & results are credible & accurate
iv. Global Acceptance of the data
Compliance to GCP:
Sponsor (Individual, company, institution or organization who initiates or manages clinical trial), Contract research organization (CRO - an independent entity that contracts with the sponsor to conduct drug development services on the sponsor’s behalf), investigator (person responsible for conduction of the trial), Investigator Site (where the trial is carried on) and Ethics Review Board (ERB) - all have to comply with the GCP guidelines. After the regulatory body and ERB approval clinical trial is initiated. Protocol adherence, proper recording in case report form, notification of the different adverse events within the time frame, declaration of principal investigator’s name and address, name of the ethics committee and approval status, regulatory clearance obtained from the Drugs Controller General of India, estimated duration of trial, site(s) of study, phase of trial, brief summary, method of generat-ing randomization sequence, method of allocation concealment, and finally method of blinding and masking.
Ethics Review Board:
It is an independent body constituting of medical or scientific professionals and non-medical or nonscientific members, whose responsibility is to ensure the protection of rights, safety and well being of human subjects involved in a trial. No clinical trial is initiated at any investigator site without obtaining a written permission by the review board. This board reviews protocol of the proposed study, informed consent document and its translation in vernacular language, all clinical and non-clinical data of the investigational product, grants, study advertisement, investigator’s qualification and trial permission by DCGI/FDA. The Ethics Review Board should perform its functions according to written operating procedures, should maintain written records of its activities and minutes of its meetings, and should comply with GCP and with the applicable regulatory requirements.
Clinical Trial Stakeholders:
There are three pillars in conducting clinical trial i.e. Sponsor/CRO, Investigator and Regulators.
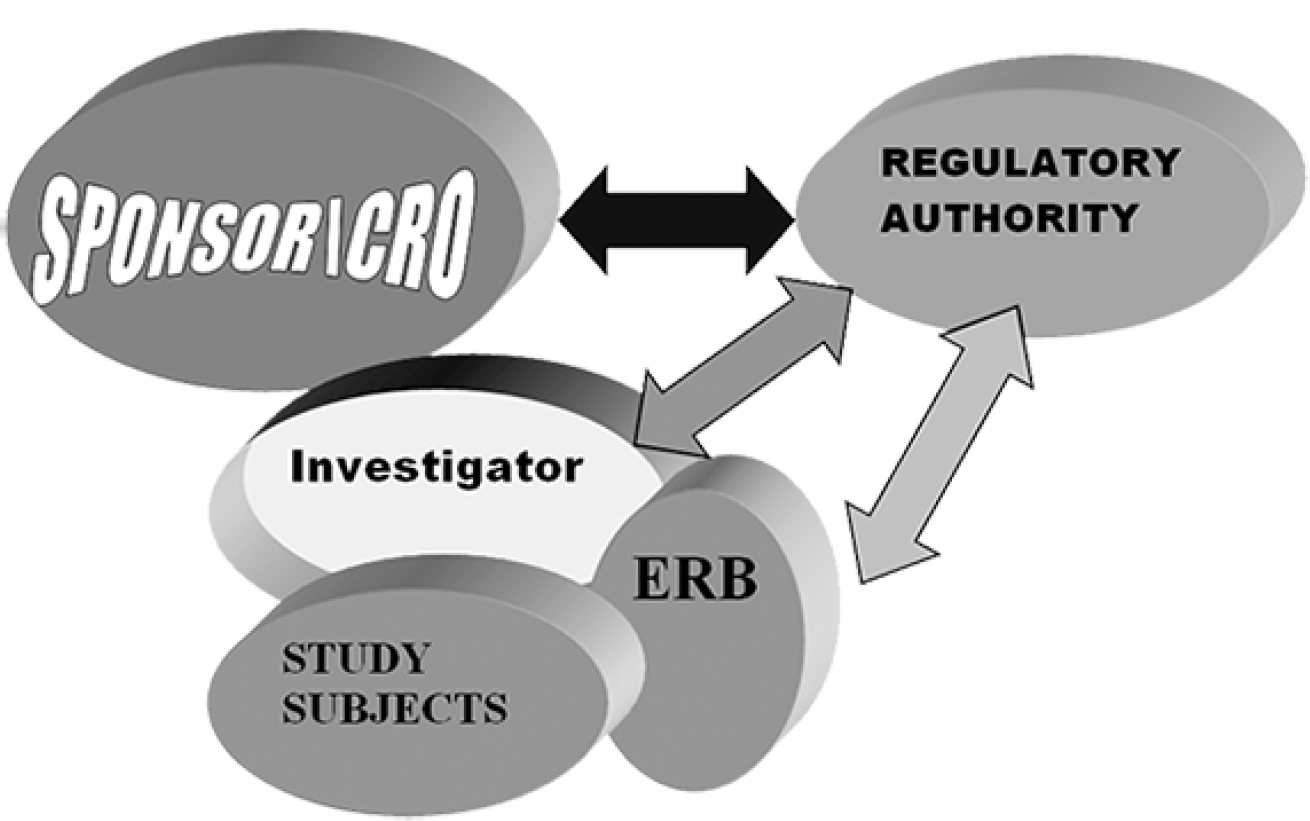
- Different Stake Holders of Clinical Traini
Conduction of clinical trials:
Different Pharmaceutical Companies, Biotechnology Companies, Contract Research Organizations (CRO), Research/Academic Institutions and Co-operative Groups can conduct clinical trial.
Ethics in Clinical trials:
India is today poised as one of the favorable destination for conducting global clinical trials due to the availability of large patient populations, skilled manpower, cost effectiveness, favorable economic environment etc. The Declaration of Helsinki (DoH) is the World Medical Association’s (WMA) best-known policy statement. The first version was adopted in 1964 and has been amended six times since, most recently at the General Assembly in October 2008. Its purpose was to provide guidance to physicians engaged in clinical research and its main focus was the responsibilities of researchers for the protection of research subjects. The advancement of medical science and the promotion of public health, although recognized as important objectives of medical research, were clearly subor-dinate to the well¬being of individual research subjects 3,4.
The Clinical Trials Registry en¬courages the registration of all clinical trials conducted in India before the enrolment of the first participant. The registry is meant to bring transparency to clinical trials conducted in India. Thus, a concerted effort on the part of the global public health community was made to push clinical trials related issues to the fore in the wake of several high-profile cases in which pharmaceutical companies were shown to be withholding information from regulators. The World Health Organization (WHO) has played a catalytic role in pushing this process forward. Though the launch of the Clinical Trials Registry marks a new chapter in the clinical trial registration process in India, there are daunting challenges ahead. Since its launch in 2007, 64 clinical trials have been registered, but there is still no legal obligation to reg¬ister. Steps are being taken to encour-age voluntary registration, including the Clinical Trials Registry workshops to which people likely to be conduct¬ WHO clinical trials - medical colleges, research institutions, state drug control¬lers, and nongovernmental organiza¬tions - are invited, but for some countries like ours, such steps are inadequate. 1
Fewer than 40 Ethics Com¬mittees in India are properly constituted and functioning, which means that the safety of the subjects of clinical trials is on the back burner, and that it is also worrying that there is no legal requirement for investigators or members of the Ethics Committees to declare a conflict of interest. Thus it is one of the serious problems given the increasing number of hospi-tals now owned by drug companies 1.
Conclusion:
Clinical trial is long, costly and complex process, requiring coordinated collaboration of a large number of individuals and groups from different departments including research, development, manufacturing, medical, regulatory, marketing and business management. It is only the successful interaction and cooperation of all professionals that can culminate in the successful registration and marketing of a new medicine. Although a set of robust guidelines is available to govern the conduct of clinical trials in any country, the conduct of clinical trial is also looked upon as an area of humanitarian concern.
Conflict of Interest:
There are no potential, perceived, or real competing and/or conflicts of interest among authors regarding the article.
REFERENCES:
- Clinical trials in India: ethical concerns. Bulletin of the World Health Organization. 2008;86(8):581-2.
- [CrossRef] [PubMed] [Google Scholar]
- The Thalidomide Tragedy. Grünenthal GmbH, Aachen 2007 Available from www.contergan.grunenthal.info. (accessed )
- [Google Scholar]
- Ethics Unit. Declaration of Helsinki. 2007 Available from www.wma.net/e/ethicsunit/helsinki.htm. (accessed )
- [Google Scholar]
- The Declaration of Helsinki and public health. Bulletin of the World Health Organization. 2008;86:650-1.
- [CrossRef] [PubMed] [Google Scholar]
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human use. 1997 Available from www.vadscorner.com/internet29.html (accessed )
- [Google Scholar]
- Ethical Guidelines for Biomedical Research on Human Participants. 2006 Available from www.icmr.nic.in/ethical_guidelines.pdf. (accessed )
- [Google Scholar]




